
Berkeley Lab Researchers Unveil Technique for Easy Comparisons of Proteins in Solution
A revolutionary X-ray analytical technique that enables researchers at a glance to identify structural similarities and differences between multiple proteins under a variety of conditions has been developed by researchers with the U.S. Department of Energy (DOE)’s Lawrence Berkeley National Laboratory (Berkeley Lab). As a demonstration, the researchers used this technique to gain valuable new insight into a protein that is a prime target for cancer chemotherapy.
“Proteins and other biological macromolecules are moving machines whose power is often derived from how their structural conformations change in response to their environment,” says Greg Hura, a scientist with Berkeley Lab’s Physical Biosciences Division. “Knowing what makes a protein change has incredible value, much like knowing that stepping on a gas pedal makes the wheels of a car spin.”
Hura led the development of what is being called a structural comparison map for use with small angle X-ray scattering (SAXS), an imaging technique for obtaining structural information about proteins and protein complexes in solution. Cynthia McMurray, a biologist with Berkeley Lab’s Life Sciences Division, provided the cancer-relevant protein used to test the new SAXS structural comparison map.
Says McMurray, “In biology, the first step in correcting a problem, such as the formation of a cancerous lesion, is understanding the conditions under which the problem arose. With the SAXS structural comparison map, we can compare multiple protein structures en masse and quickly identify areas of interest.”
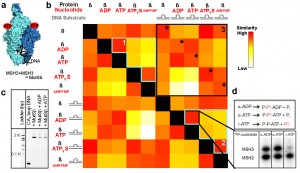
Data in this revolutionary structural comparison map is presented as a color-coded checkerboard with similarity scores displayed as gradients moving from red, indicating high, to white, indicating low, and various shades of orange and yellow in between. Image credit: Berkeley Lab (Click image to enlarge)
Hura is the lead author and McMurray one of two corresponding authors of a paper in the journal Nature Methods that describes this research. The paper is titled “Comprehensive objective maps of macromolecular conformations by quantitative SAXS analysis.” Also a corresponding author is John Tainer, who holds joint appointments with Berkeley Lab’s Life Sciences Division and the Scripps Research Institute. The other authors are Helen Budworth, Kevin Dyer, Robert Rambo and Michal Hammel.
In perhaps no other area of science does the maxim “function follows form” hold more true than for proteins and protein complexes. The structural conformations created by the folding, twisting and turning of a protein’s amino acid chain can allow or prevent the protein from doing what it’s supposed to do and this can mean the difference between a healthy and an unhealthy cell. A protein can assume multiple distinct conformational states as it undergoes various chemical processes such as phosphorylation, nucleotide or ligand binding, ATP hydrolysis or the formation of complexes.
The most widely used technique for determining a protein’s structure remains crystallography, but many proteins and protein complexes can’t be crystalized. Furthermore, though precise, crystallography is a low-throughput process that can only capture one conformational state at a time. Enter SAXS, a high-throughput technique that can image any protein or protein complex in solution under any condition, and provide nanoscale resolution for distinguishing and characterizing the different conformational states that flexible biological macromolecules such as proteins can assume.
“With SAXS, there are relatively few restraints on conditions, construction, concentration or solution chemistry,” Hura says. “However, analytical methods have not kept pace with the hardware. While there are many factors that may induce a protein to undergo structural changes, these factors are difficult to predict. Our structural comparison map technique gives us a high-throughput screening capability. The combination of SAXS and our maps allows us to highlight those factors that make the biggest difference in structural conformations. We’re also able to track trends and identify intermediate states and other factors that shift equilibrium from one structure to another.”

Greg Hura, Helen Budworth and Cynthia McMurray, shown here at the Advanced Light Source, developed a structural comparison map for SAXS imaging and tested it on a chemotherapeutic target protein. (Photo by Roy Kaltschmidt, Berkeley Lab)
The data in a structural comparison map is presented in the form of a color-coded checkerboard with similarity scores displayed as gradients moving from red, indicating high, to white, indicating low, and various shades of orange and yellow in between.
“With structural comparison maps, I can immediately see which structures under which conditions are the same and which are not,” says McMurray. “The maps provide both structural and chemical information and enable us to identify those conformations we should be looking at.”
To test the structural conformation map technique, co-author Budworth, a member of McMurray’s research group, prepared samples of a protein known as MutSß, an inviting chemotherapeutic target because of its ability to remove problematic DNA that can lead to cancer and other genetic mutations.
“MutSß is a heterodimer whose two macromolecules undergo an ordered series of nucleotide-dependent steps to initiate DNA repair,” Budworth says. “Each discrete nucleotide-bound state is a conformational state decision point that primes the next pathway step. A mechanistic understanding of these steps is crucial to learning how cells avoid mutation.”
Says McMurray, “Initially this was a very big puzzle because MutSß had no crystal structure, nor could we take a look at any one conformational state and say this is good or this is bad. The structural conformation maps allowed us to characterize the different conformational states individually and then compare them to one another. We discovered that DNA has surprisingly little impact on MutSß conformational structures, a fact that was not evident from biochemical measurements, but obvious when examining the maps.”
From the SAXS imaging and structural conformation map analysis, McMurray and her group believe that DNA is sculpted to the protein conformation and that nucleotide-binding drives MutSß conformational changes. This, they say, holds implications for future cancer therapies.
The MutSß samples were subjected to SAXS at the SIBYLS beamline of Berkeley Lab’s Advanced Light Source, a synchrotron that generates premier beams of X-ray and ultraviolet light for scientific research. The acronym SIBYLS stands for Structurally Integrated Biology for Life Sciences. The beamline is maintained by Berkeley Lab’s Life Sciences Division under the direction of corresponding author Tainer.
Says Tainer, “The structural comparison map technique is a big step forward in the development of tools that will help biologists use the full potential of the awesome throughput we expect to achieve with the next generation of light sources.”
This research was supported by funds from the DOE Office of Science and from the National Institutes of Health.
– By Lynn Yarris
*Source: Berkeley Lab