
Mycobacterium tuberculosis, the bacterium that causes tuberculosis may have an Achilles’ heel: It needs a particular enzyme to survive. Inhibiting that enzyme’s function, researchers have shown, will kill the bacteria, pointing toward a design strategy for new TB drugs.
PROVIDENCE, R.I. [Brown University] — To stay ahead in the race against drug-resistant infections, scientists constantly search for and exploit vulnerabilities in deadly bacteria. Now, researchers from Brown and the Massachusetts Institute of Technology have used a novel compound to exploit an Achilles’ heel in the bacterium that causes tuberculosis.
In a series of laboratory experiments, the researchers have shown that it is possible to kill Mycobacterium tuberculosis by inhibiting ClpP, a cellular enzyme that is not targeted by any antibacterial drug on the market. The work is preliminary, but the researchers are hopeful it could point the way to new drugs to treat tuberculosis and other infections that are becoming resistant to traditional antibiotics.
“ClpP has emerged over the last decade or so as a potential drug target in bacteria because they require it to either live or to cause disease,” said Jason Sello, associate professor of chemistry at Brown, who led the research. “Our findings indicate that chemical inhibition of the essential ClpP enzyme in Mycobacterium tuberculosis is a viable strategy in anti-tuberculosis drug development.”
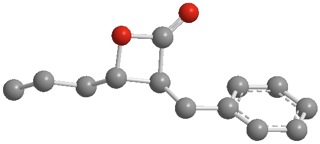
A small molecule packs a punch. β-lactone 7, a small molecule that inhibits the ClpP enzyme in bacteria, may be important in development of new anti-bacterial drugs. The bacterium that causes tuberculosis cannot survive when ClpP is inhibited. Image credit: Sello lab/Brown University
A paper describing the research has been accepted by the journal ACS Chemical Biology and published online.
The ClpP (pronounced “clip-P”) enzyme is sometimes referred to as “the garbage disposal of the bacterial cell,” Sello said. It degrades and eliminates proteins inside the cell that are misfolded, damaged, or have outlived their usefulness. While ClpP is dispensable in most bacteria, previous research has shown that it is absolutely essential for the viability of M. tuberculosis. When the gene that makes the protein is knocked out of the bacterium’s genome, the organism cannot survive.
“If you can inhibit the function of the enzyme with a small molecule, then you can kill the bacterium,” Sello said. “When we began our work, the validity of this approach hadn’t been demonstrated.”
Sello and Corey Compton, a fourth-year graduate student in his group, set out to do so in the lab. Previous research had found that small molecules called β-lactones inhibit the ClpP protein in other bacteria, suggesting that they might do the same in M. tuberculosis.
To find out, the researchers synthesized 14 β-lactones, each with a slightly different chemical structure. They tested the effect of those molecules against Mycobacterium smegmatis, a close relative of the tuberculosis bacterium often used in the lab because it does not cause disease. Of the 14 molecules, four were shown to kill M. smegmatis in petri dish cultures. To see if the compounds could also kill M. smegmatis’s deadly cousin, Sello and Compton sent them to the Institute for Tuberculosis Research at the University of Illinois for further testing. There, they found that the compounds were indeed effective in killing M. tuberculosis as well. The most potent compound they identified — β-lactone 7 — had a structure that was different from all other known inhibitors of ClpP. The potency of this compound is in the same range as streptomycin, a clinically used tuberculosis drug.
The next step was to confirm that the β-lactone 7 and the other compounds were killing the bacteria by reacting with and inhibiting ClpP, as the team expected. To do that, Sello and his team marked the β-lactones with a kind of chemical handle that allowed them to retrieve any proteins with which the molecules had reacted. By putting these tagged β-lactones in a culture of M. smegmatis (the tuberculosis stand-in), the researchers were able to show that they did in fact bind to the two proteins that construct the ClpP enzyme.
“These results were extremely gratifying,” Compton said. “The compounds not only interacted with ClpP but they did so quite selectively.”

Graduate student Corey Compton, left, and Jason Sello synthesized 14 β-lactones. Four of them killed a close relative of the TB bacterium. β-lactone 7 was the most potent. Image credit: Brown University
The final step was proving that the reaction between the β-lactones and ClpP actually resulted in the inhibition of the enzyme’s activity. For that, Sello turned to Robert Sauer at MIT, an expert on the ClpP enzyme. Sauer and Karl Schmitz, a postdoctoral fellow in his research group, had developed an in vitro assay in which the activity of ClpP could be measured in a test tube. Using this assay, Schmitz and Sauer found that when ClpP was treated with β-lactones the activity of the enzyme was inhibited.
Taken together, the results suggest that molecules that inhibit ClpP could be an important addition to the drug arsenal for fighting tuberculosis.
“Our data validate ClpP as a viable, antibacterial drug target,” said Sello. “We have a high degree of confidence that inactivating ClpP will inhibit the growth of Mycobacterium tuberculosis. In principle, a pharmaceutical company could develop new tuberculosis drugs by using the structure of β-lactone 7 as a starting point or by using ClpP inhibition as a design strategy”.
The work was funded by Brown University and the National Institutes of Health (GM-101988).
– By Kevin Stacey
*Source: Brown University