
Researchers were able to improve walking and prolong survival in a mouse model by tweaking potassium levels
By boosting the ability of a specific type of cell to absorb potassium in the brain, UCLA researchers were able to improve walking and prolong survival in a mouse model of Huntington’s disease.
Their findings, published March 30 in the online edition of the journal Nature Neuroscience, could point to new drug targets for treating the devastating disease, which strikes one in every 20,000 Americans.
Huntington’s disease is passed from parent to child through a mutation in the huntingtin gene. By killing brain cells called neurons, the disorder gradually deprives patients of their ability to walk, speak, swallow, breathe and think clearly. No cure exists, and patients with aggressive cases can die in as little as 10 years.
Artistic rendering of astrocyte. Image credit: University of California
The laboratories of Baljit Khakh, a UCLA professor of physiology and neurobiology, and Michael Sofroniew, a UCLA professor of neurobiology, teamed up at the David Geffen School of Medicine at UCLA to unravel the role that astrocytes — large, star-shaped cells found in the brain and spinal cord — play in Huntington’s.
“Astrocytes appear in the brain in equal numbers to neurons yet haven’t been closely studied,” Khakh said. “They enable neurons to signal each other by maintaining an optimal chemical environment outside the cells. We used two mouse models to explore whether astrocytes behave differently during Huntington’s disease.”
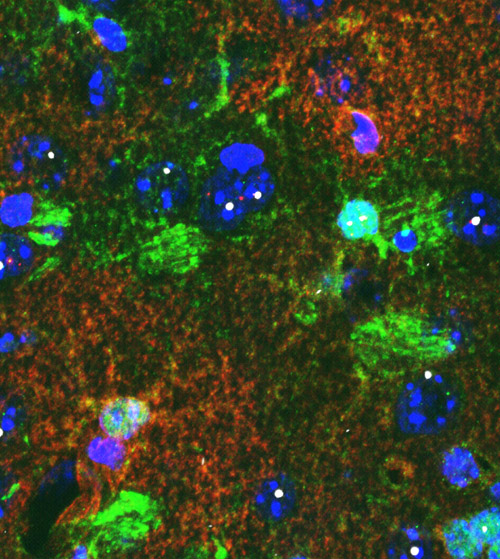
Astrocytes in brain tissue. Brain tissue from a mouse shows star-shaped astrocytes (green). Cells (blue) containing mutant protein (white) display lower levels of a potassium-regulating protein (red). Image credit: University of California
The first model mimicked an aggressive, early-onset type of the disorder, while the second imitated a slow-developing version. Khakh and Sofroniew, both members of the UCLA Brain Research Institute, examined how the huntingtin mutation influenced astrocytes in the brain. In particular, they looked at astrocytes’ interaction with a type of neuron that plays a central role in coordinating movement.
One key finding stood out from the data.

Michael Sofroniew, M.D., Ph.D. Image credit: University of California
In both models, astrocytes with the mutant gene showed a measurable drop in Kir4.1, a protein that allows the astrocyte to take in potassium through the cell membrane. This left too much potassium outside the cell, disrupting the chemical balance and increasing the nearby neurons’ “excitability,” or capacity to fire.
“We suspect that the gene mutation contributes to Huntington’s disease by reducing Kir4.1 levels in the astrocytes,” Sofroniew said. “This, in turn, reduces the cell’s uptake of potassium. When excess potassium pools around neurons, they grow oversensitive and fire too easily, disrupting nerve-cell function and ultimately the body’s ability to move properly. This may contribute to the jerky motions common to Huntington’s disease.”
To test their hypothesis, the scientists explored what would happen if they artificially increased Kir4.1 levels inside the astrocytes. The results proved striking.
“Boosting Kir4.1 in the astrocytes improved the mice’s ability to walk properly,” Khakh said. “We were surprised to see the length and width of the mouse’s stride return to more normal levels. This was an unexpected discovery.”
“Our work breaks new ground by showing that disrupting astrocyte function leads to the disruption of neuron function in a mouse model of Huntington’s disease,” Sofroniew said. “Our findings suggest that therapeutic targets exist for the disorder beyond neurons.”

Baljit Khakh, Ph.D. Image credit: University of California
While the results shed important light on one of the mechanisms behind Huntington’s disease, the findings also offer more general implications, according to the authors.
“We’re really excited that astrocytes can potentially be exploited for new drug treatments,” said Khakh. “Astrocyte dysfunction also may be involved in other neurological diseases beyond Huntington’s.”
The UCLA team’s next step will be to tease out the mechanism that reduces Kir4.1 levels and illuminate how this alters neuronal networks.
The study was supported by the CHDI Foundation, the National Institute of Mental Health and the National Institute of Neurological Disorders and Stroke.
Khakh and Sofroniew’s co-authors included UCLA’s Xiaoping Tong, Yan Ao, Guido Faas, Ji Xu, Martin Haustein, Mark Anderson and Istvan Mody and the University of Alabama–Birmingham’s Sinifunanya Nwaobi and Michelle Olsen.
– By Elaine Schmidt
*Source: University of California